Beginner’s Guide to Generative Design for Small Molecules
This guide introduces the principles of generative design and how it's helping researchers create fit-for-purpose small molecules.
7 min read
July 14th, 2025
Last updated: July 18th, 2025

Introduction
The way we discover new drugs is undergoing a fundamental shift. For decades, drug discovery has largely relied on screening existing molecules by searching the chemical space based on what we already have. But the question has evolved with generative design.
Instead of asking "What should we test next?", it encourages us to ask "What should we make next?", thereby opening up a whole new world of opportunities.
This blog post captures the core principles of computational generative design of small molecules. It walks you through how modern algorithms build, evaluate, and navigate chemical space to generate novel molecular candidates.
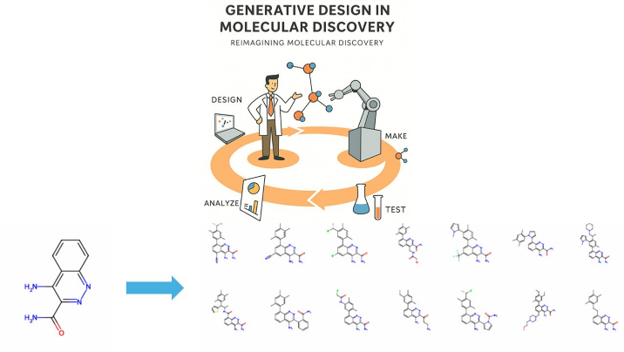
Figure 1: An example of the set of molecules that can be generated from a single organic compound. Source: Yassir Boulaamane
What is Generative Design in Drug Discovery?
Generative design refers to the use of computational tools to create novel molecules from scratch. It’s not merely a tool for enumeration.
It is a creative process that blends chemical knowledge, machine learning, and optimization to explore new territories of chemical space.
In the classical drug discovery pipeline, we begin with known compounds and screen them for biological activity. This strategy, while effective in some contexts, has limitations, particularly when it comes to underexplored targets, IP challenges, or novel disease areas.
Generative design inverts the model. It allows us to design molecules that do not just ‘exist’ but are purposefully created to meet desired criteria such as potency, bioavailability, novelty, synthetic accessibility, and more.
This mindset also aligns perfectly with the iterative design-make-test-analyze (DMTA) cycle. Instead of passively screening what's available, generative design injects intelligence into each step of the cycle. The focus shifts from libraries of known compounds to strategic construction of new ones. This enables the rational design of molecules for targets previously deemed ‘undruggable.’
Generative Design Starts with Molecular Representation
To design molecules computationally, we must first determine how to represent them in a digital form. This challenge lies at the heart of cheminformatics, the discipline dedicated to encoding, manipulating, and managing chemical structures and data.
How Machine Learning Reads Chemical Structures - Read more here!
2D format
One of the most basic molecular representation formats is the 2D depiction. While these are intuitive for humans, they’re not computationally useful. For meaningful operations such as generation, scoring, and transformation, we move toward graph-based representations.
Molecular graphs
A molecular graph is a mathematical structure where atoms are represented as nodes and bonds as edges. This abstraction allows for efficient traversal and modification, making it the backbone of most generative algorithms.
These graphs are often serialized into SMILES (Simplified Molecular Input Line Entry System) strings. SMILES is a compact, linear representation that encodes molecular structure in text format.
It is especially popular because it integrates well with machine learning, databases, and generative models. However, SMILES strings are not unique by default, which necessitates a process known as canonicalization.
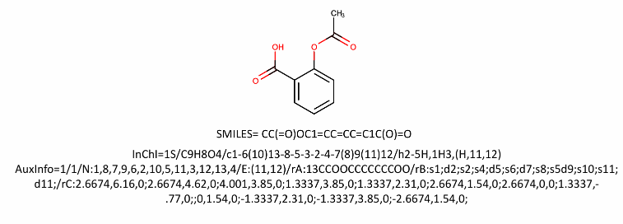
Figure 2: Structure and chemical identifiers of aspirin (acetylsalicylic acid). Source: Yassir Boulaamane
Another widely used format is InChI (International Chemical Identifier), which generates a unique identifier for each molecule, accounting for stereochemistry and tautomeric forms.
InChIs are ideal for deduplication and archiving but are less flexible for generative tasks.
3D formats
When we move beyond static structures, we step into 3D molecular representations. These include ball-and-stick models with explicit hydrogens and interatomic distances, useful for simulations and docking. At the highest level of detail, mesh-based 3D representations include electron density, enabling accurate property prediction, albeit at high computational cost.
The same representational principles apply to chemical reactions. Reactions are encoded using formats like reaction SMILES, SMARTS, SMIRKS, or condensed reaction graphs. These representations allow us to generalize transformations which are essential for reaction-based generative approaches.
Learn how to apply generative models to real-world drug design challenges.
Molecular Construction for Building New Molecules
Generative design methods are ultimately about construction. How do we generate new chemical structures that are novel, meaningful, and synthesizable?
There are three principal strategies for molecular construction: atom-based, fragment-based, and reaction-based. Each comes with its own trade-offs between control and feasibility.
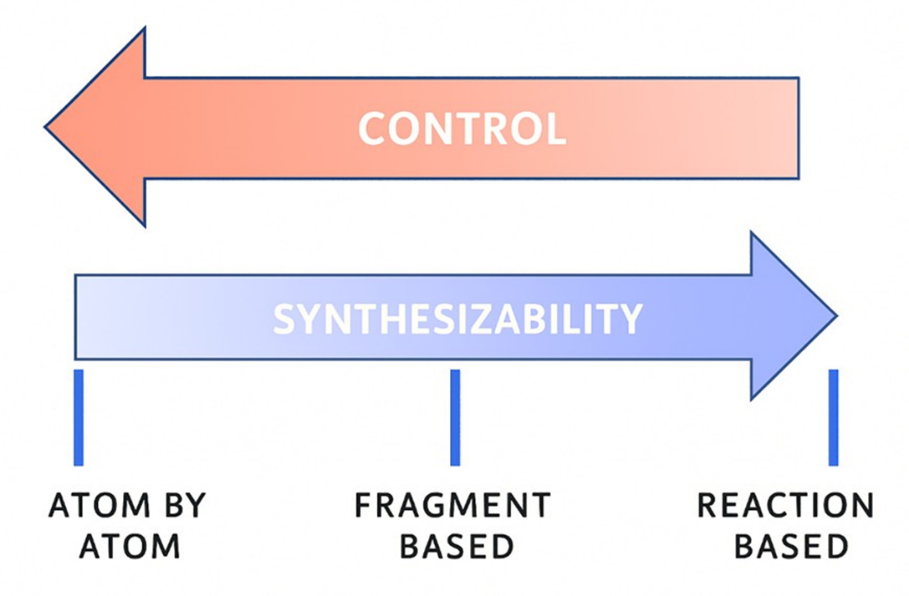
Figure 3: Three principal strategies for molecular construction Source: Yassir Boulaamane
Atom-based construction
Atom-by-atom design offers the highest level of control. It allows for arbitrary placement of atoms and bonds, enabling exploration of novel regions of chemical space that may never have been synthesized.
However, with this freedom comes a cost.
Molecules constructed atom-by-atom may violate valency rules or be synthetically inaccessible. Managing valence constraints, stereochemistry, and ring strain becomes increasingly complex. Furthermore, it’s often necessary to apply multiple transformations in sequence to correct valency issues or remove implausible substructures.
Despite these challenges, atom-based methods are powerful, especially when paired with strong scoring functions and post-generation filters.
Fragment-based construction
Fragment-based methods strike a balance between control and realism. Here, molecules are assembled from pre-defined fragments typically extracted from known compounds or vendor libraries. Fragments may include ring systems, linkers, and side chains.
This approach benefits from synthetic plausibility. Since the fragments come from real molecules, they’re more likely to be chemically valid. However, choosing the right fragments and deciding how to join them remains non-trivial. Fragment growing, linking, and merging are common tactics used to generate new scaffolds from existing pieces.
The major advantage is that these methods reduce the complexity of molecular generation while still offering creative flexibility, especially when the fragment space is carefully curated.
Reaction-based construction
Reaction-based approaches offer the most direct path from “in silico” design to “in vitro” synthesis. Instead of drawing molecules from scratch, this strategy builds new compounds by applying known chemical reactions to known building blocks.
These reactions are typically encoded as generalized rules, often in SMIRKS format. For instance, an amide coupling reaction can be applied to any compatible amine and acid to generate a product. Because the transformations mirror real synthetic routes, the output is inherently more ‘makeable’ than structures from other approaches.
This method also integrates well with electronic lab notebooks and automation pipelines. It forms a natural loop where chemists create molecules, log reactions, and those same reaction patterns feed back into generative systems.
Want to go deeper? Take the full course on Generative AI for Drug Discovery and start designing molecules with confidence.
Scoring for Turning Molecules into Meaningful Numbers
Construction is only half the story! Once we've generated molecules, we need to evaluate them. This is where scoring comes into play.
The goal of molecular scoring is to assign a numerical value to a molecule that reflects its desirability. This value is what guides optimization, whether in reinforcement learning, genetic algorithms, or simple hill climbing.
Scoring methods fall into two broad categories: explicit and implicit.
Explicit scoring
Explicit scoring is rule-based and quantifiable. It includes properties such as molecular weight, LogP, number of hydrogen bond donors/acceptors, and docking scores. Advanced techniques include free energy perturbation, molecular dynamics, and machine learning-based property prediction.
These scores are reproducible and objective, making them ideal for algorithmic optimization.
Implicit scoring
Implicit scoring captures subjective factors such as ‘what a chemist likes about a molecule’.
This may involve perceived tractability, synthetic accessibility, or project-specific preferences. Though hard to formalize, implicit scoring often reflects the nuanced reality of drug development.
In practice, the best generative systems blend both. For instance, a molecule may score highly on explicit metrics but be rejected due to a known PAINS motif or synthetic infeasibility, both caught through implicit filters.
Multi-objective optimization frameworks, like Pareto front analysis, are useful for managing trade-offs. For example, increasing potency might decrease solubility. Pareto-optimal solutions are those where no objective can be improved without worsening another.
Filtering also plays a key role. Filters like Lipinski’s Rule of Five or PAINS substructure checks allow rapid elimination of unfit candidates before deeper evaluation.
Search Algorithms for Navigating Chemical Space
Chemical space is large, estimated at over 10⁶⁰ possible molecules. Generative design doesn't just involve construction and scoring. It also requires an efficient search function.
How do we intelligently explore the vast chemical space without getting lost?
Search algorithms guide the generative process by deciding which structures to explore next. They can be broadly categorized as deterministic, stochastic, or hybrid.
Deterministic search
Deterministic strategies follow fixed rules. Systematic enumeration is one such method: apply all possible transformations and evaluate the resulting compounds. While exhaustive, this quickly becomes computationally intractable.
Greedy algorithms, such as hill climbing, improve molecule quality step-by-step but can get trapped in local optima.
Stochastic and hybrid methods
Stochastic approaches, like Monte Carlo sampling, introduce randomness to explore chemical space more broadly. Epsilon-greedy strategies combine exploration and exploitation, randomly choosing a transformation with a certain probability.
One powerful hybrid method is simulated annealing. It begins with high randomness (exploration) and gradually becomes more focused (exploitation), mimicking how chemists operate early vs. late in an optimization campaign.
These techniques allow generative tools to escape local traps and discover better solutions.
Generative Design for Small Molecules Using Machine Learning
Many state-of-the-art generative approaches now integrate machine learning. These models learn structure-property relationships and use them to guide molecule generation.
Popular architectures include:
| Model Architecture | Description |
|---|---|
| Recurrent Neural Networks (RNNs) | Generate SMILES strings character by character. |
| Variational Autoencoders (VAEs) | Encode molecules into a latent space, enabling interpolation and optimization. |
| Generative Adversarial Networks (GANs) | Generate realistic molecules by pitting a generator against a discriminator. |
| Reinforcement Learning (RL) agents | Learn to construct molecules by maximizing reward functions tied to desired properties. |
These models leverage vast datasets, like ChEMBL or PubChem, and can generate libraries orders of magnitude larger than traditional enumeration.
Towards Autonomous Drug Design
Generative design is evolving rapidly. Today, we use it to accelerate ideation and expand chemical space. But the future is even more ambitious.
Imagine a fully autonomous design loop. A machine defines the hypothesis, generates candidate molecules, passes them to a robotic synthesis platform, and feeds results back into the model, all without human intervention.
While we’re not there yet, the groundwork is being laid.
Together, let's make molecules that matter!
Neovarsity is a Berlin-based deep tech skills platform. We build industry-driven, cohort-based programs in collaboration with world-class experts to prepare talent and teams to solve problems in areas with real-world impact.
For questions, contact Catherine at [email protected]. Stay tuned for more updates and insights.
Follow us on LinkedIn and join the conversation using #FutureThroughDeepTech.
Design novel small molecules with generative AI
Develop and use generative models to deploy complete pipelines for drug discovery.
- Represent molecules using graphs, SMILES, and 3D encodings for model-ready inputs
- Generate, optimize, and evaluate compounds using deep generative architectures
- Create a capstone portfolio of real-world AI applications in small molecule design